phasedIBD
Fast and robust identity-by-descent inference with the templated positional Burrows-Wheeler transform
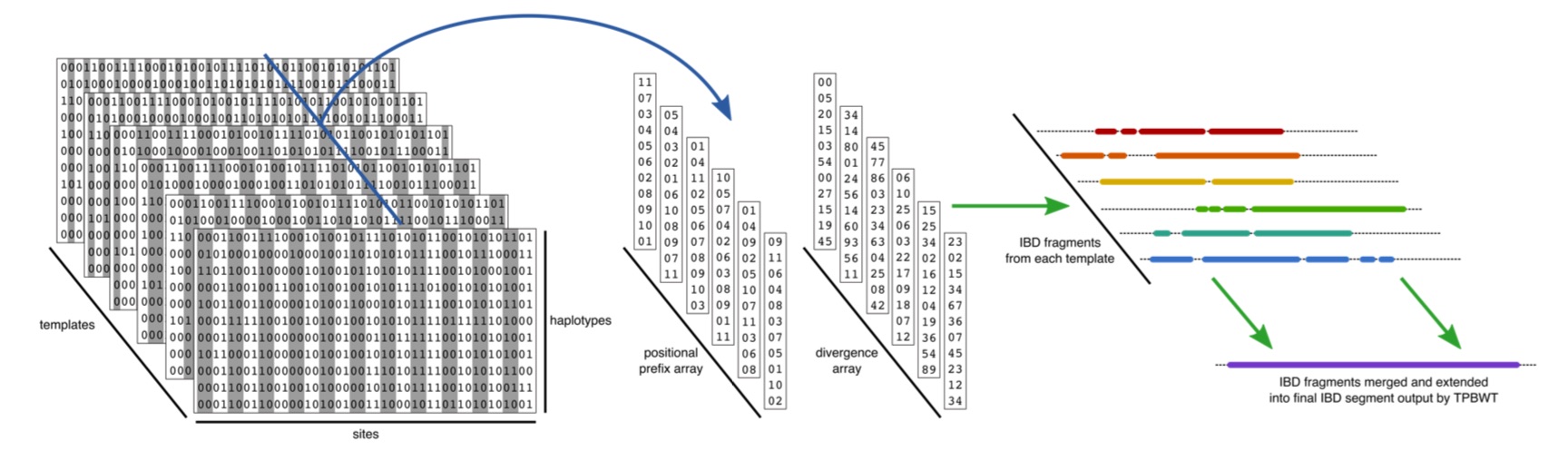
Estimating the genomic location and length of identical-by-descent (IBD) segments among individuals is a crucial step in many genetic analyses. However, the exponential growth in the size of biobank and direct-to-consumer (DTC) genetic data sets makes accurate IBD inference a significant computational challenge. The templated positional Burrows-Wheeler transform (TPBWT) makes fast and efficient IBD estimates robust to haplotype and phasing errors. Our in-sample and out-of-sample TPBWT-based IBD inference algorithms have the computational efficiency to run on massive-scale datasets with millions of samples. Furthermore the software can produce TPBWT-compressed haplotypes in a binary file format that enables fast and efficient out-of-sample IBD computes against very large cohort panels.
Our software implementation of the TPBWT is freely available in the code repository https://github.com/23andMe/phasedibd.
Citation:
Freyman, W.A., McManus, K.F., Shringarpure, S.S., Jewett, E.M., and A. Auton. 2021. Fast and robust identity-by-descent inference with the templated positional Burrows-Wheeler transform. Molecular Biology and Evolution 38(5) 2131–2151.
[preprint]
[journal]
[pdf]

RevBayes
I was a regular contributor to the software RevBayes, where I worked on ancestral state reconstruction functionality and developed both continuous-time Markov chain and state-dependent speciation and extinction models of character evolution. I'm also interested in the inference of reticulate evolution, and have implemented coalescent models to estimate ancient introgression/hybridization events over a phylogeny.
I coathored a couple RevBayes tutorials:
- State-dependent speciation and extinction models
Analyses using BiSSE, HiSSE, and ClaSSE.
- Chromosome evolution models
Analyses using ChromEvol, BiChroM, ChromoSSE and related models.

SUMAC 2.0
8/2016: SUMAC version 2.0 is significantly faster than previous versions due to the use of a new clustering algorithm.
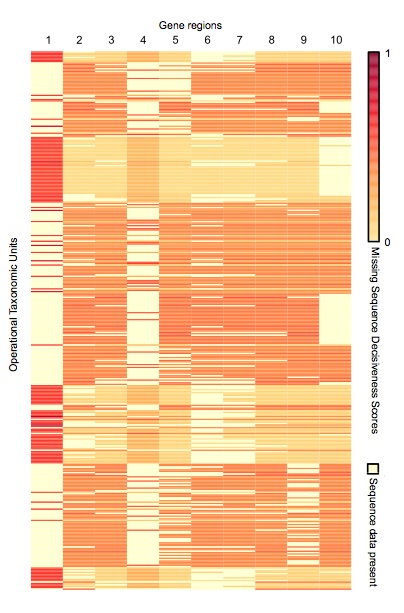
Supermatrix Constructor (SUMAC) is a tool to data-mine GenBank, construct phylogenetic supermatrices, and assess the decisiveness of a matrix given the pattern of missing sequence data. SUMAC calculates a novel metric, Missing Sequence Decisiveness Scores (MSDS), which measure how much each individual missing sequence contributes to the decisiveness of the matrix. MSDS can be used to compare supermatrices and prioritize the acquisition of new sequence data.
SUMAC constructs supermatrices either through an exploratory clustering of all GenBank sequences within a taxonomic group, or by using guide sequences to build homologous clusters in a more targeted manner. SUMAC will assemble supermatrices for any taxonomic group recognized in GenBank, and is optimized to run on multicore processors by utilizing multiple parallel processes. SUMAC is implemented as a Python package that can run as a stand-alone command line program, or its modules and objects can be incorporated within other programs.
SUMAC works on Linux/OSX (not MS Windows), and is available at https://github.com/wf8/sumac under the open source GPLv3 license.
Citation:
Freyman, W.A. 2015. SUMAC: constructing phylogenetic supermatrices and assessing partially decisive taxon coverage. Evolutionary Bioinformatics 2015:11 263-266
[html]
[pdf]



Universal FQA Calculator
Floristic Quality Assessments (FQAs) are metrics of ecological quality developed by Swink and Wilhelm in 1994. FQAs are calculated using coefficients of conservatism assigned to individual plant species based on their endemism to a certain habitat and tolerance to disturbance.
Widely used by state and federal agencies as well as conservation non-profits to monitor and assess natural areas, FQA databases have been developed for much of the United States. However, all previous FQA computer programs only calculated FQA for a single region or habitat, and for many regions FQA must be calculated by hand. This web-based FQA calculator enables the user to choose from any existing FQA database, and as new FQA databases are developed they can be uploaded into the site. Users can also compare coefficients across databases. The site has a responsive user interface that can be used in the field on mobile devices. This project was developed with support from Openlands.
Source code: https://github.com/wf8/universalFQA
Citation:
Freyman, W.A., L.A. Masters, and S. Packard. 2016. The Universal Floristic Quality Assessment (FQA) Calculator: an online tool for ecological assessment and monitoring. Methods in Ecology and Evolution 7(3): 380–383 [html] [pdf]

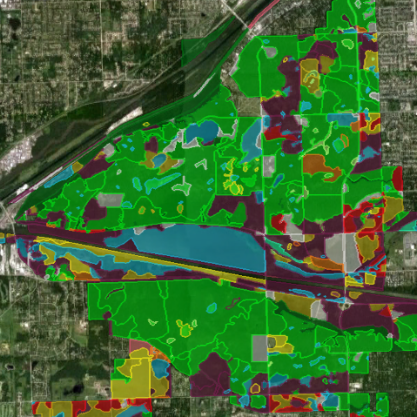
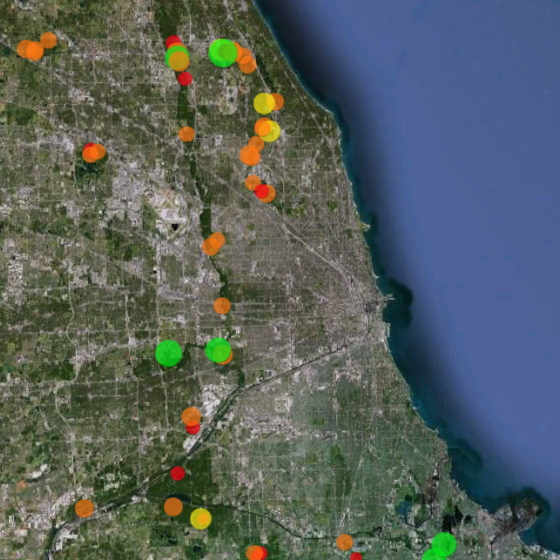
Restoration Map
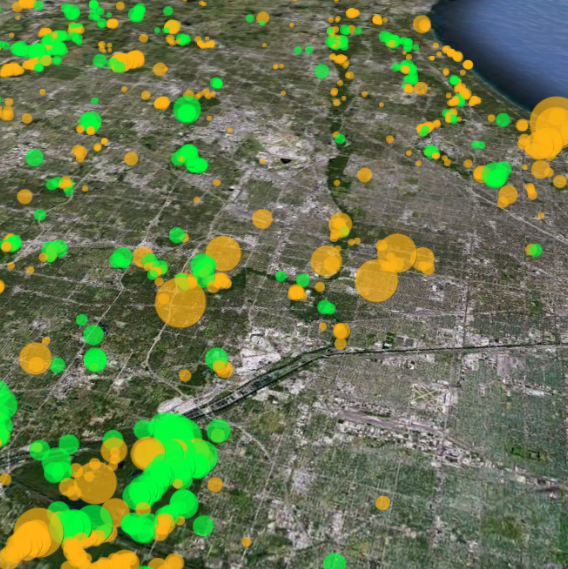
Restoration Map is an open-source and free web-based application to help plan, implement, and assess ecological restoration projects within Chicago Wilderness natural areas. It is designed to encourage collaboration and improve communication among stakeholders including the landowner agencies, volunteers, restoration contractors, interns, and partner conservation organizations.
The map's users have entered over 1800 map layers representing 13 years of management work and experimentation in Chicago-area restoration projects. By integrating long term monitoring data from the Bird Conservation Network (eBird), the Calling Frog Survey, and other sources, Restoration Map provides feedback on the effects of restoration work, therefore utilizing an adaptive management approach.
Restoration Map has been developed with the support of the National Audubon Society using all free and open-source software. The concept was inspired by Stephen Packard's stewardship of the beautiful Somme Prairie Grove. The map is built on PHP and MySQL, and uses the Google Earth plugin.
Learn more at http://restorationmap.org
Source code: https://github.com/wf8/restorationmap
Citation:
Freyman, W.A. and K.A. Glennemeier. 2014. Restoration Map: a web-based tool for spatial and participatory adaptive management of ecological restoration projects. Ecological Restoration (32-1) [html] [pdf]