Research
The evolution of life has long been understood to be a branching process through time, with all extant species representing twigs on the tree of life. In the past 20 years, evolutionary biology has made enormous progress by mathematically modeling this branching process using the framework of phylogenetic theory. Currently, phylogenetic theory faces two major challenges that my research tackles: (1) estimating the effect of trait evolution on macroevolutionary diversification rates; and (2) inferring the reticulate evolutionary history of lineages that are simultaneously undergoing incomplete lineage sorting and processes such as hybridization or introgression.



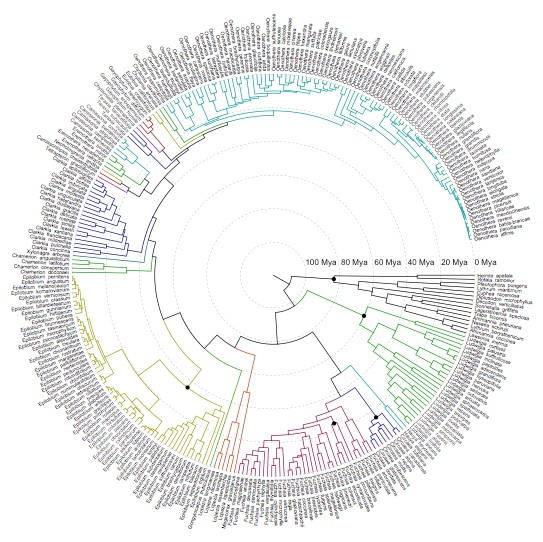
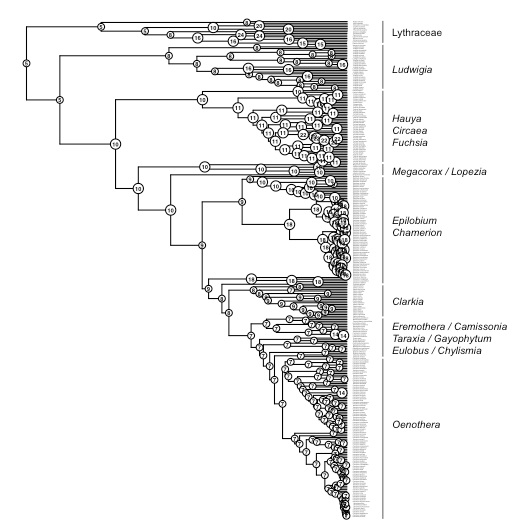
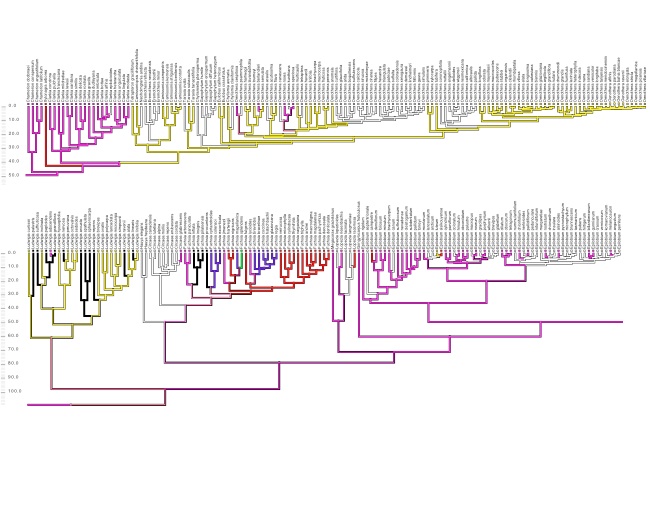
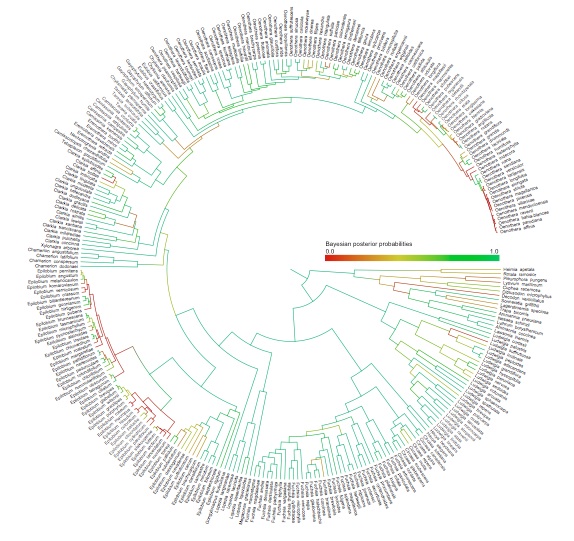

Detecting introgression in the presence of incomplete lineage sorting: phylogenomics and hybridization in Chylismia (Onagraceae)
Increasingly, hybridization is understood to be an important evolutionary process in a substantial portion of many plant and animal groups. One explanation for the extent of hybridization can be found in G. Ledyard Stebbins’ 1959 hypothesis that hybridization acts as an evolutionary catalyst by increasing the adaptive variation within lineages. However, most current phylogenetic models assume bifurcating trees and are inadequate to test hypotheses involving hybridization and other reticulate patterns. To address this, I've developed a composite likelihood approach that estimates ancient introgression and hybridization events over a phylogeny. The model combines elements of both coalescent theory and phylogenetics by calculating the joint posterior probability of a coalescent model of ABBA and BABA incongruent gene tree histories simultaneously with a phylogenetic molecular evolution model conditioned on a population tree.
The plant genus Chylismia (Onagraceae) is an ideal study group to empirically test these new models, based on the strong foundation of cytogenetic, biosystematic, and descriptive data detailed in Peter Raven’s monograph. Chylismia consists of 35 taxa and includes a homogamic (diploid) hybrid complex of 14 species distributed throughout the harsh habitats of the Sonoran Desert and Great Basin of North America. Raven hypothesized that the members of Chylismia diverged during a rapid radiation resulting from the increasing aridity of the Great Basin region following the uplift of the Sierra Nevada. He further inferred that the complex of highly variable and hybridizing Chylismia species is the result of repeated vicariance and recontact due to pluvial lake cycles during the Pleistocene.
Motivated both by the challenges of phylogenetic network inference and by Stebbins’ 1959 hypothesis of hybridization as a catalyst for evolution, this research incorporates both empirical and theoretical work: (1) I'm using next-generation sequencing and genomic datasets to study the evolutionary consequences of introgression and historical biogeography across the hybridizing Chylismia species, (2) applying both empirical Chylismia datasets and simulated datasets to evaluate the new model described above, and (3) quantifying the genomic structural changes that accompany reticulate evolution and their functional significance in Chylismia using whole chloroplast genomes and RAD markers from across the nuclear genome.
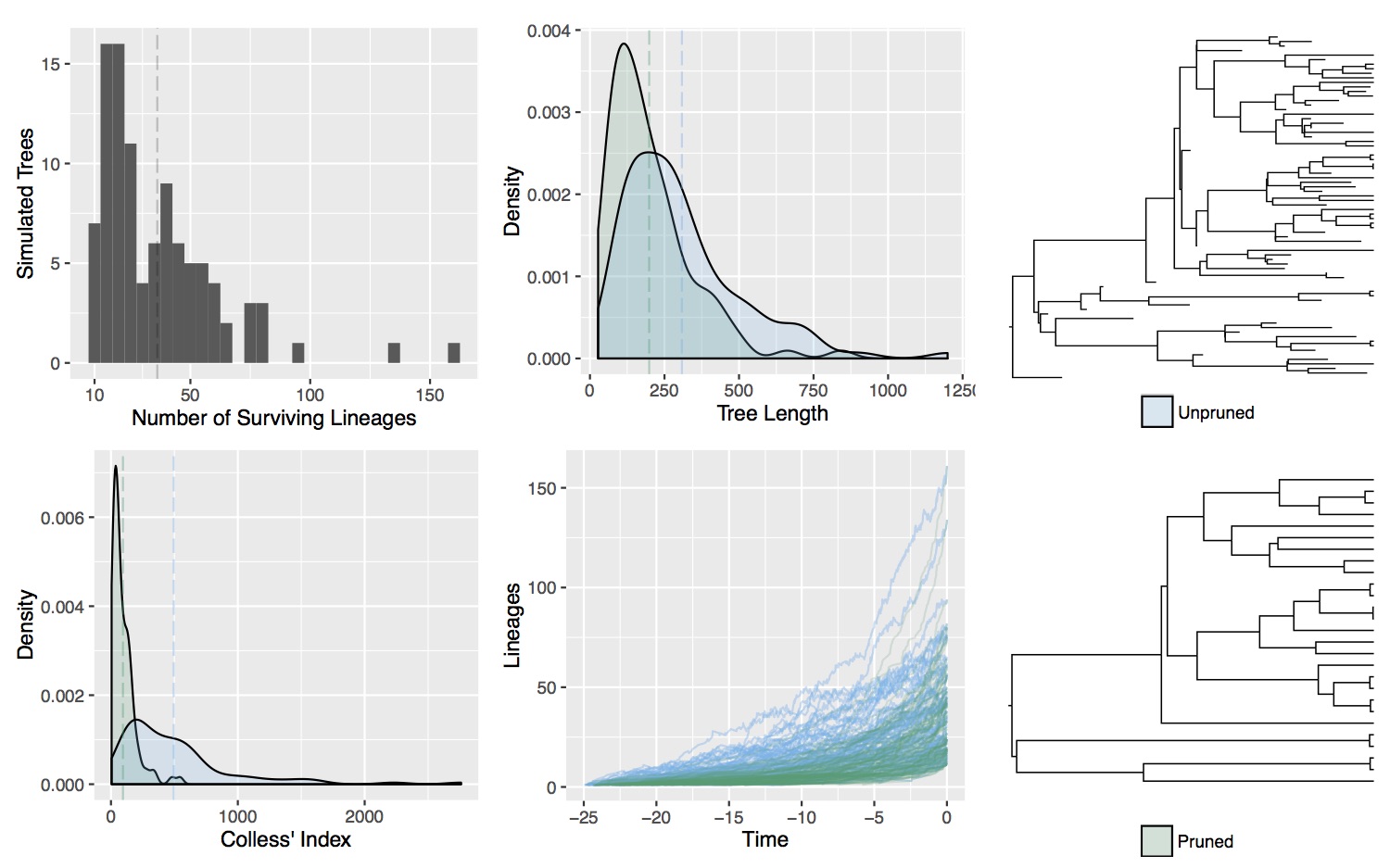
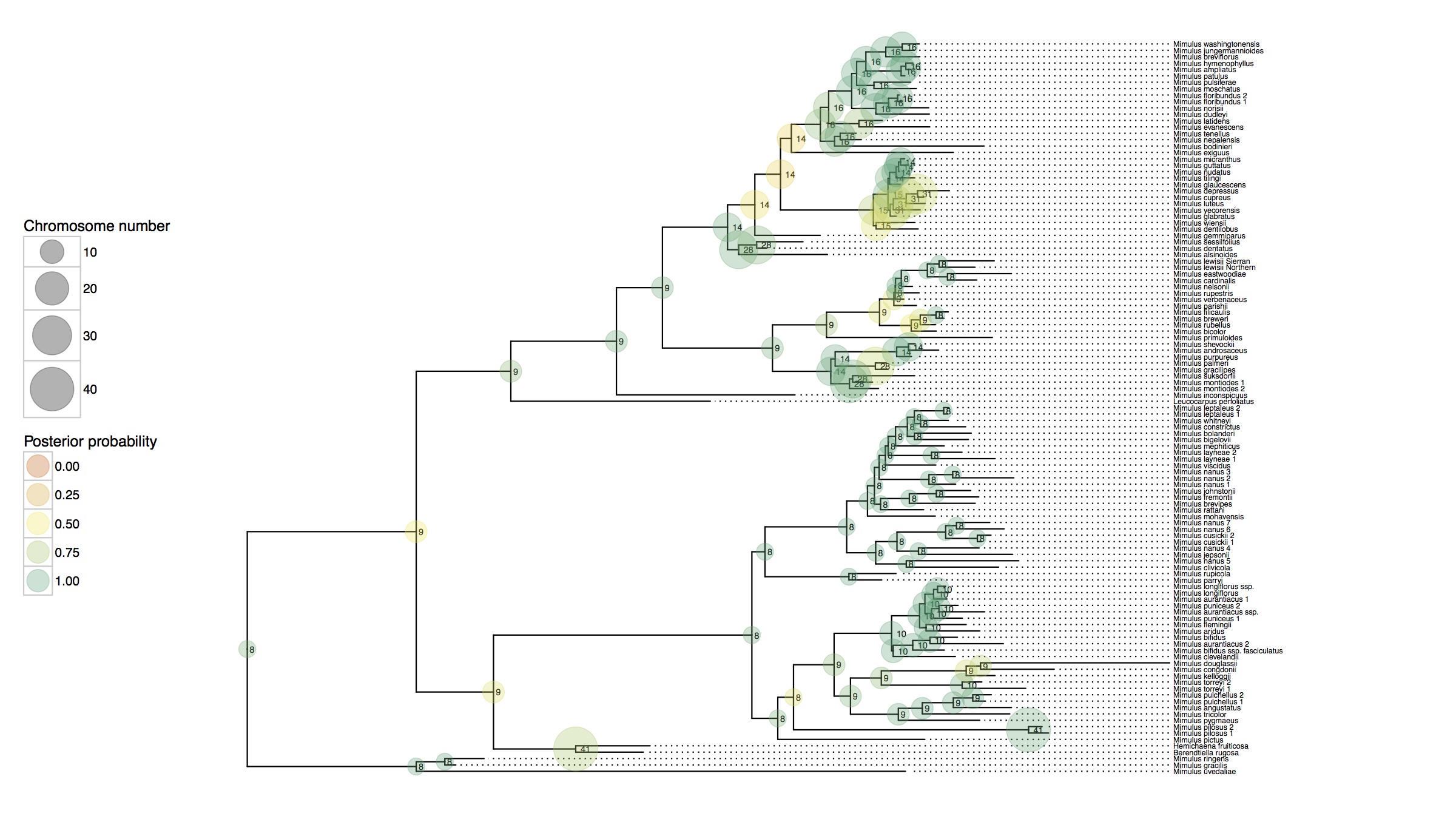
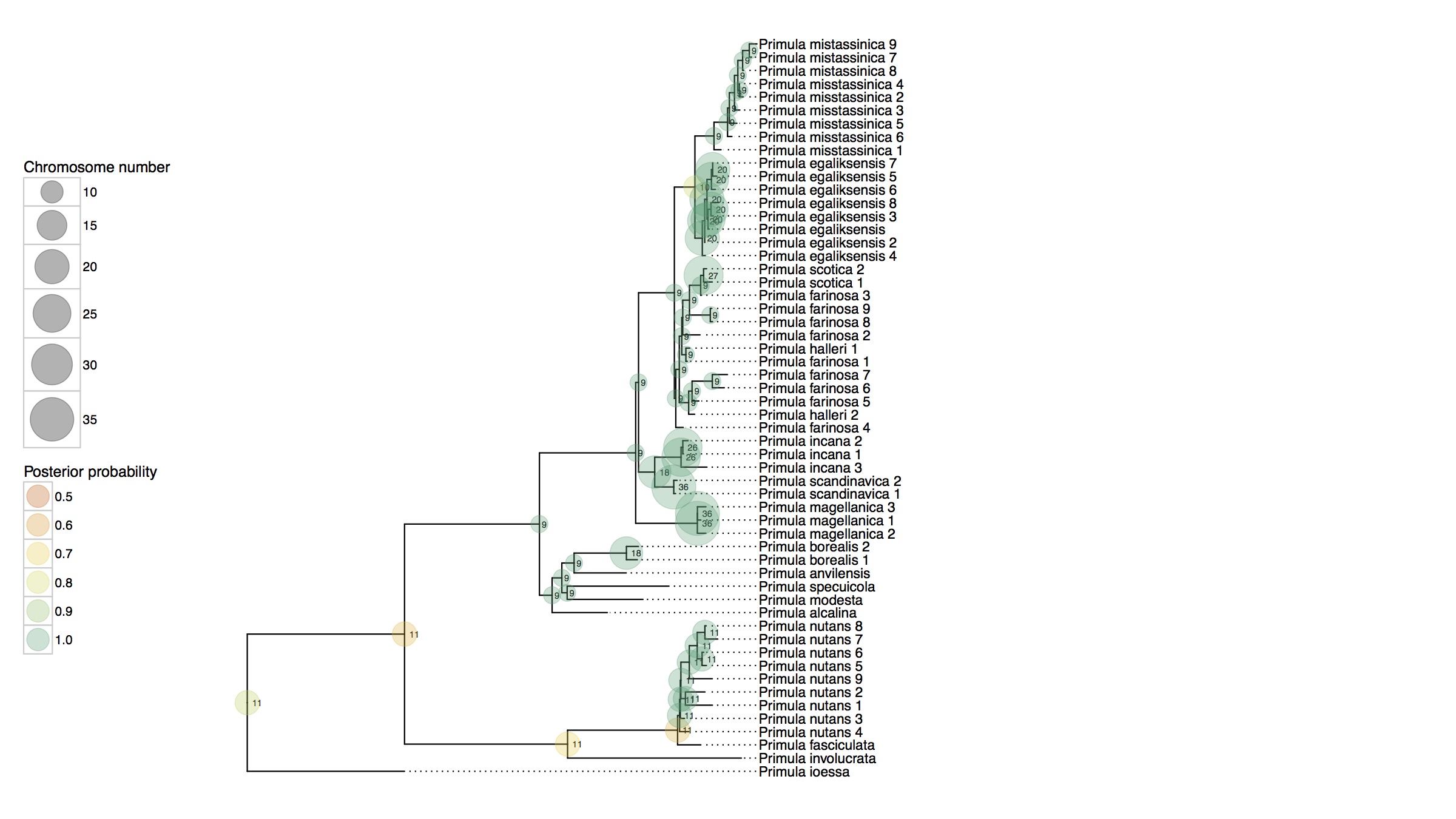
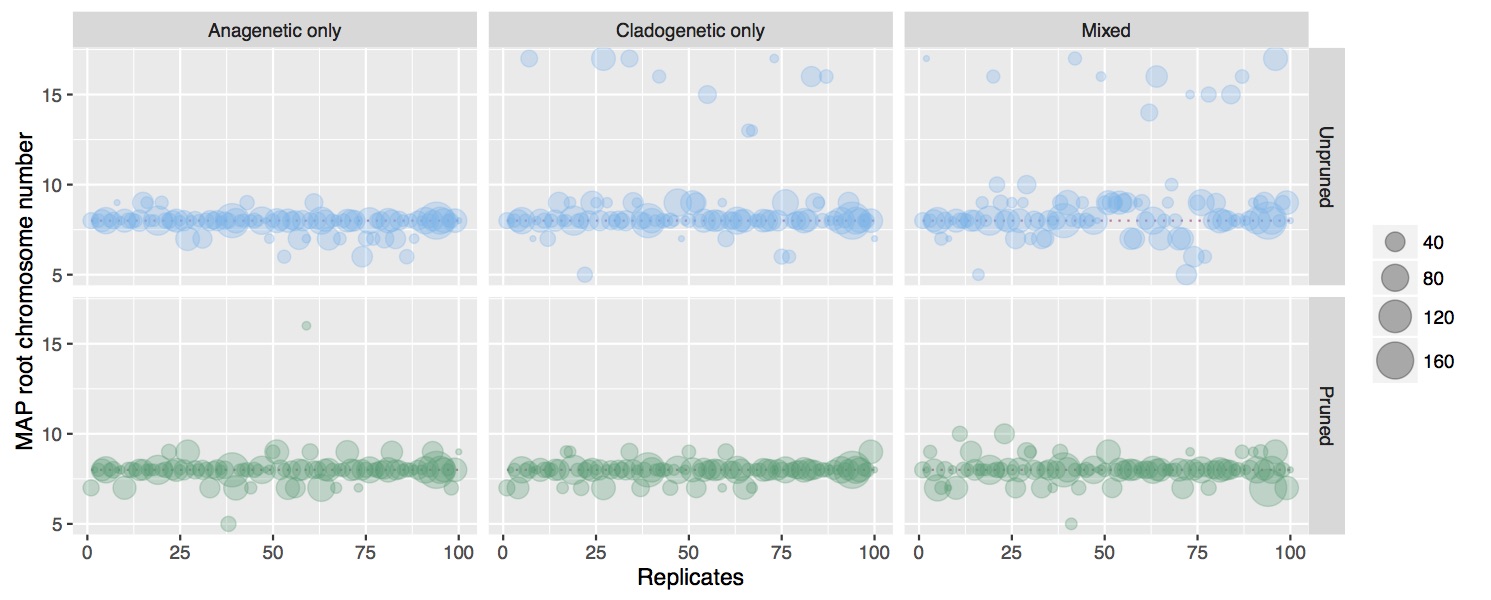
Cladogenetic and anagenetic models of chromosome number evolution: model averaging using reversible jump MCMC
The fundamental role that chromosomal changes play in evolution was confirmed in Dobzhansky's groundbreaking Genetics and the Origin of Species (1937), which unified Mendelian genetics and Darwinian natural selection into a single coherent theory of evolution. "Chromosomal changes are one of the mainsprings of evolution," Dobzhansky asserted, and chromosomal rearrangements such as deletions, duplications, inversions, and translocations could help explain the formation of reproductive isolation and lineage diversification. The recent development of probabilistic models of chromosome evolution in the groundbreaking work by Mayrose et al. (2010) have enabled the inference of ancestral chromosome numbers over molecular phylogenies and generated new interest in studying the role of chromosome changes in evolution. However, these models assume all changes occur anagenetically (along branches), and do not model events that are specifically cladogenetic and may be expected if chromosomal changes result in reproductive isolation.
In this research I've developed a class of models of chromosome number evolution that incorporate both anagenetic and cladogenetic change. The motivation in developing these new models is to determine the mode of chromosome number evolution; is chromosome evolution occurring primarily within lineages, primarily at lineage splitting, or in clade-specific combinations of both? By identifying how much of the pattern of chromosome number evolution is explained by anagenetic versus cladogenetic change, we can help uncover how much of a role chromosomal changes play in speciation.